EXHIBIT 99.1
Published on
Exhibit 99.1
BUSINESS
Overview
We are a vertically integrated medical device company focused on developing and commercializing innovative medical devices to treat people with obesity. Our initial product offering is the Obalon® Balloon System, the first and only U.S. Food and Drug Administration, or FDA, approved swallowable, gas-filled intragastric balloon designed to facilitate progressive and sustained weight loss as an adjunct to a moderate intensity diet and behavioral program for patients with obesity. We believe the Obalon Balloon System offers patients and physicians benefits over prior weight loss devices including, but not limited to: a favorable safety profile, improved patient tolerability and comfort, progressive weight loss with durable results, simple and convenient placement, and potentially attractive economics for patients and physicians.
The Obalon Balloon System is FDA approved for temporary use to facilitate weight loss in adults with obesity having a body mass index, or BMI, of 30 to 40, or approximately 30 to 100 pounds overweight, who have failed to lose weight through diet and exercise. The system is intended to be used as an adjunct to a moderate intensity diet and behavior modification program. All balloons must be removed in six months after the first balloon is placed. We believe the Obalon Balloon System has provides patients and physicians with a cost-effective, reversible and repeatable weight loss solution in an outpatient setting, without altering patient anatomy or requiring surgery.
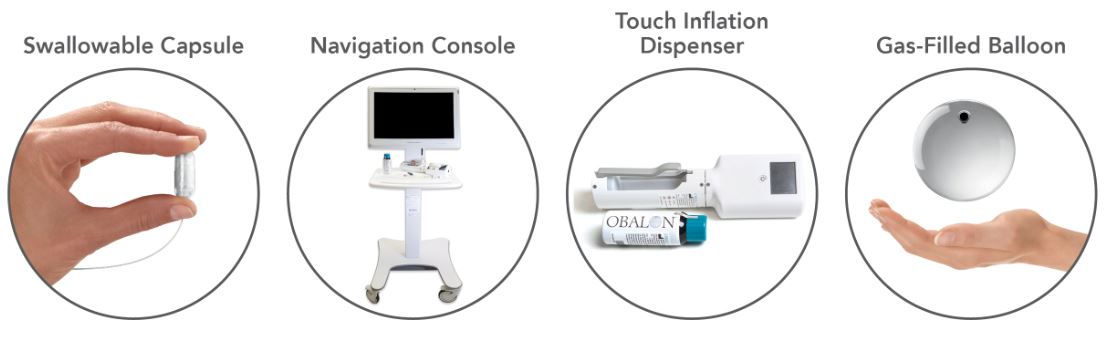
The current generation of our Obalon Balloon System consists of a swallowable capsule that contains an inflatable balloon attached to a microcatheter; the Navigation System console, which is a combination of hardware and software used to track and display the location of the balloon during placement; the Obalon Touch Inflation Dispenser, which is a semi-automated, hand-held inflation device used to inflate the balloon once it is placed; and a disposable canister filled with our proprietary mixture of gas. Placement of a balloon typically occurs in less than 10 minutes and can be accomplished in an outpatient setting. Patients receive a total of three balloons over the course of eight to 12 weeks and all balloons are removed six months after the first balloon is placed.
The Obalon Balloon System has demonstrated progressive weight loss with durable results. In our SMART trial, patients in the Obalon treatment group lost, on average, approximately twice as much body weight as patients in the sham-control group. In addition, patients in the Obalon treatment group showed, on average, progressive weight loss over the entire six-month balloon treatment period, and maintained, on average, 89.5% of the weight loss six months after balloon removal. In December 2018, we analyzed data from our commercial registry on more than 1,300 patients at 108 treatment sites. For those patients who received three balloons and at least 20 weeks of therapy, the average weight loss was 21.7 pounds, resulting in 9.9% reduction in total body weight and a 3.5 point decrease in BMI compared to baseline values. Of note, the top quartile of those patients lost an average of 39 pounds, resulting in a 16.8% reduction in total body weight and a 6.2 point decrease in BMI compared to baseline values. Furthermore, in May 2019, additional data was presented, which included additional data analysis from our commercial registry of 1,411 total patients from 143 treatment sites in the United States. In this larger data set, for those patients receiving three balloons and at least 20 weeks of therapy, the average weight loss was 21.7 pounds, resulting in a 10.2% reduction in total body weight. Of note, 50.7% of patients lost 10% or more total body weight and 77.9% lost 5% or more total body weight. Weight loss in the first months of therapy was the largest predictor of success.
We commenced U.S. commercialization of our prior generation Obalon balloon system in January 2017. In February 2019, we commercialized our Obalon Touch Inflation Dispenser and our Obalon Navigation System, which together are intended to make balloon placement more reliable, safer, easier and less expensive. The Obalon Navigation System is designed to eliminate the need to use x-ray technology when placing the Obalon balloon.
Historically we have sold our products directly to physicians, who would then sell weight loss treatment packages to their patients that included our balloon therapy, dietary counseling and balloon removal on a non-reimbursed, self-pay basis. We are
1
currently undergoing a process to fundamentally change our commercialization efforts, which commenced with the elimination of our direct sales force in connection with an overall workforce reduction in April 2019. Concurrent with that reduction, we transitioned to a centralized customer support model through which we sell to existing physicians that are using our balloons or new physicians that contact us directly to acquire our system and balloons and provide marketing and clinical support to those physicians. Marketing support may include media assets and media purchasing support, access to our call center, leads generated from our Find-A-Doc locater and leads generated from our digital and off-line marketing efforts. In addition, rather than focusing on selling directly to physicians, we intend to establish company-owned or managed Obalon-branded retail centers. Under this new model, we plan to either directly employ the physicians or contract for their services, and we will be directly responsible for marketing and patient acquisition. We would also provide the staff, equipment and support services necessary for patients to receive Obalon balloon therapy from licensed physicians. We believe this model will contribute to standardization of both quality of care and patient pricing and provide us greater operational and financial control of our business. We plan to launch our first Obalon branded retail center before year end 2019.
Obesity is one of the largest, costly and underserved disease states globally. In adults, the disease is linked to several co-morbidities, including hypertension, type 2 diabetes, high blood pressure, certain cancers and other chronic conditions. The national medical care costs of obesity-related illness in adults, including out of pocket expenses, third-party payer expenses and Medicaid, were estimated to be up to $210 billion in 2008.
Current treatment alternatives for obese patients begin with lifestyle modification, such as diet and exercise. If this alternative fails to produce the desired results, physicians may prescribe pharmaceutical therapies, typically to obese or overweight patients with a lower body mass index, or BMI. Although pharmaceutical therapies have been effective in assisting with weight loss, they are often associated with safety risks and negative side effects that may limit patient compliance. In obese patients with a higher BMI, physicians may pursue aggressive surgical treatments, such as gastric bypass and gastric banding. These procedures promote weight loss by surgically restricting the stomach’s capacity and outlet size; however, they present substantial side effects and carry short- and long-term safety risks that have limited adoption. Intragastric balloons were first introduced in 2015 and represent a relatively new category of treatment for weight loss in the United States. We believe traditional liquid-filled intragastric balloons suffer from limitations that have impeded their adoption, including their rate of serious adverse device events, a lack of comfort and tolerability, a limited ability to provide progressive and sustained weight loss and an inconvenient placement procedure.
We believe the Obalon Balloon System addresses many of these limitations and provides the foundation for an important, growing and sustainable treatment for weight loss.
The Obesity Epidemic
Obesity has been identified by the U.S. Surgeon General as an epidemic and a significant threat to the quality of life in the United States. Based on results from the 2013-2014 National Health and Nutrition Examination Survey, it is estimated that more than 88 million adults in the United States were obese, defined as a BMI of 30 or greater, of which approximately 18 million were considered extremely obese with a BMI of 40 or greater, and an approximately 76 million adults in the United States were overweight, defined as a BMI between 25 and 29.9. Research sponsored by the Centers for Disease Control and Prevention, or CDC, suggests that if current obesity rates persist, half of the U.S. population will be obese by 2030. Obesity is also a significant health problem outside of the United States. The number of obese adults worldwide has nearly tripled since 1975, and the World Health Organization estimates that more than 650 million adults were obese and more than 1.9 billion were overweight in 2016.
The CDC has identified obesity as a leading cause of preventable death in the United States, and it is one of the leading causes of chronic diseases both worldwide and in the United States. Obesity-related disorders, known as comorbidities, include cardiovascular diseases, diabetes, musculoskeletal disorders and some cancers. The national medical care costs of obesity-
2
related illness in adults, including out-of-pocket expenses, third-party payer expenses and Medicaid, were estimated to be up to $210 billion in 2008. Furthermore, in 2014 the annual global economic impact of obesity was estimated to be $2 trillion.
We expect the obesity epidemic among adults to continue to grow worldwide given the excess caloric intake of highly-processed, fatty foods, increasingly sedentary lifestyles and a growing prevalence of obesity among children and adolescents. Despite the growing public interest in the obesity epidemic and the significant medical and economic repercussions associated with the disease, there remains a significant unmet need for more effective treatments.
Current Treatments and Limitations
Current treatment alternatives for patients who are obese and overweight begin with lifestyle modification, such as diet and exercise. If this course of treatment fails to produce the desired results, physicians may prescribe pharmaceutical therapies, and in patients with more severe obesity, physicians may pursue aggressive surgical treatments, such as gastric bypass and gastric banding. These approaches are associated with safety concerns, lifestyle impact and ease of use, cost and compliance issues that have limited their adoption. Additionally, some patients may seek to address the symptoms of weight-gain through the use of aesthetic products, certain of which have been approved for individuals with a BMI of 30 or less. These aesthetic products are also not indicated for weight loss.
Lifestyle Modification
Lifestyle modification, which includes diet, exercise and behavior modification, is usually prescribed as an initial treatment for a patient who is obese or overweight and is typically prescribed in all obesity management approaches. However, lifestyle modification alone has generally been ineffective in producing sustainable weight loss in patients with obesity due to inability to comply with the modifications over an extended period. Many studies have shown that a significant majority of dieters will regain lost weight and many will gain more than they originally lost.
Pharmaceutical Therapy
Several pharmaceutical products have been approved by the FDA for obesity in the United States. Pharmaceutical therapy often represents a first option in the treatment of patients with obesity that have failed to achieve weight loss goals through lifestyle modifications alone. Pharmaceutical therapy can have limited effectiveness due to patient non-compliance. Additionally, pharmaceutical therapy may carry significant safety risks and negative side effects, such as adverse gastrointestinal, cardiovascular and central nervous system issues, some of which are serious or life threatening.
Bariatric Surgery
Bariatric surgery is a treatment option generally reserved for cases of severe obesity, or patients with a BMI in excess of 40. The most common forms of bariatric surgery, gastric bypass and sleeve gastrectomy, promote weight loss by surgically restricting the stomach’s capacity and outlet size. Gastric bypass also affects weight loss by restricting the body’s ability to absorb nutrients. While largely effective, these procedures are generally invasive, expensive for the patient and irreversible. Bariatric surgery patients are generally required to make significant postoperative lifestyle changes, including strict dietary changes, vitamin supplementation and long-term medical follow-up programs. Side effects of bariatric surgery include a high rate of re-operation, nausea, vomiting, dumping syndrome, dehydration, dental problems and other issues.
Recently Developed Treatment Alternatives
Given the shortcomings and limitations of the existing treatment alternatives, new medical procedures have been recently introduced in an attempt to address the gap in care between pharmaceutical treatment and invasive surgical procedures. These new procedures include: neuroblocking therapy, aspiration therapy, hydrogel technology, gastric emptying technology, and liquid-filled intragastric balloons. Neuroblocking therapy involves a surgical procedure in which a neuromodulation device is implanted in the body and used to block electrical signals from the stomach to the brain. By blocking those signals, the device
3
attempts to control the patient’s feelings of hunger. Aspiration therapy involves a surgical procedure in which a feeding tube is implanted in the abdomen in order to remove food from the stomach before calories are absorbed into the body. We believe high costs, procedural complications and the risk of serious adverse device events, or SADEs, may limit their adoption.
Intragastric balloons are a type of space-occupying device placed in the stomach in order to cause a sensation of fullness. Currently marketed traditional balloons are large, liquid-filled silicone devices that are placed in the stomach endoscopically, under anesthesia, for a treatment period of up to six months. Following treatment, the balloons are removed in a second endoscopic procedure. Approved traditional liquid-filled intragastric balloons in the United States are the ReShape Duo Balloon and the ORBERA Balloon. While generally effective in delivering weight loss, these traditional liquid-filled intragastric balloons have been accompanied by a number of limitations that have impeded their adoption, including: high rate of SADEs, lack of comfort and tolerability, limited ability to provide progressive and sustained weight loss, and inconvenient placement procedure.
FDA recently cleared Plenity, a device developed by Gelesis based on hydrogel technology. The device is designed to expand in the stomach by absorbing water to create the feeling of satiety. It is similar to pharmacotherapy in that it is highly dependent on patient compliance to ensure effectiveness of the product as the product is self-administered and is approved for consumption of three capsules to be consumed with water before both lunch and dinner. FDA has approved BAROnova's transpyloric shuttle, a non-surgical, non-pharmacologic device to induce weight loss by slowing gastric emptying by blocking the pyloric channel such that food remains in the stomach longer. Limitations for the BAROnova product are similar to those experienced with liquid-filled balloons including endoscopic placement and endoscopic removal and an increased rate of device and procedure-related adverse events.
Our Solution
We have developed our Obalon Balloon System to overcome the limitations of prior devices intended to treat weight loss, including traditional liquid-filled intragastric balloons. Based on our clinical data and commercial experiences, we believe the Obalon Balloon System provides the following benefits to our patients and their physicians:
• | Favorable safety profile. In our pivotal SMART trial, only one of 336 patients (0.3%) that received our Obalon balloon experienced a SADE, and in data presented at the American Society for Metabolic and Bariatric Surgery Meeting from our first year of commercial experience, only two of 1,343 patients (0.14%) that received our Obalon balloon experienced a SADE. As of December 31, 2018, the rate of SADEs reported to us in commercial use remains similar with that experienced in either the pivotal SMART trial or the data from our first year of commercial experience. |
• | Improved patient tolerability and comfort. The Obalon balloon is inflated with a proprietary mix of gas. This creates a light, buoyant balloon that floats at the top of the stomach instead of sinking to the bottom of the stomach like a traditional liquid-filled intragastric balloon. Further, the Obalon Balloon System consists of three separate 250cc balloons placed individually over a three-month period to progressively add volume. We believe these design elements have the potential to improve patient comfort and tolerability of our Obalon balloon. |
• | Progressive weight loss with durable results. In our pivotal SMART trial, patients in the Obalon treatment group lost, on average, approximately twice as much body weight as patients in the sham-control group. In addition, patients in the Obalon treatment group showed, on average, progressive weight loss over the balloon treatment period, which we believe was attributable to the individual placement of three separate Obalon balloons over the treatment period. Subsequent data analysis at 12 months also showed that, on average, 89.5% of the weight loss was maintained six months after balloon removal. In December 2018, we analyzed data from our commercial registry on more than 1,300 patients at 108 treatment sites. For those patients who received three balloons and at least 20 weeks of therapy, the average weight loss was 21.7 pounds, resulting in 9.9% reduction in total body weight. Furthermore, in May 2019, |
4
additional data from the registry was presented to include a total of 1,411 total patients from 143 treatment sites in the United States. In this expanded data set, for those patients receiving three balloons and at least 20 weeks of therapy, the average weight loss was 21.7 pounds, resulting in a 10.2% reduction in total body weight.
• | Simple and convenient placement. The Obalon balloon is placed without anesthesia or an endoscopy through a swallowable capsule that dissolves in the stomach and releases the balloon. These unique features allow patients the flexibility to receive the Obalon balloon discreetly in an outpatient setting. Placement typically occurs in less than ten minutes and can be scheduled in the morning before work, during a lunch break or in the evening. Treated patients can return promptly to their normal daily activities. The balloons are removed endoscopically under light, conscious sedation six months after the first balloon placement. Recently approved new products, the Obalon Navigation System and Obalon Touch Inflation Dispenser, further improves convenience of placement. |
Our Strategy
Our objective is to be the leading provider of medical devices for the non-surgical treatment of persons who are obese. The key elements of our strategy are to:
• | Launch and pursue a company-owned or managed Obalon branded retail center commercialization model. Historically we sold our products directly to physicians, who would then sell weight loss treatment packages to their patients that included our balloon therapy, dietary counseling and removal on a non-reimbursed, self-pay basis. We are currently undergoing a process to fundamentally change our commercialization efforts, which commenced with the elimination of our direct sales force in connection with an overall workforce reduction in April 2019. Concurrent with that reduction, we transitioned to a centralized customer support model through which we sell to existing physicians that are using our balloons or new physicians that contact us directly to acquire our system and balloons and provide marketing and clinical support to those physicians. In addition, rather than focusing on selling directly to physicians, we intend to establish company-owned or managed Obalon branded retail centers. Under this model, we plan to either directly employ the physicians or contract for their services, and we will be directly responsible for marketing and patient acquisition activities. We will also provide or contract the staff, equipment and support services necessary for patients to receive Obalon balloon therapy from licensed physicians. We believe this model will allow us to standardize both quality of care and patient pricing, and provide us greater operational and financial control of our business. We plan to launch our first Obalon branded retail center before year end 2019 and, assuming the first center is successful, anticipate opening additional centers over the course of 2020. |
• | Continue to drive patient awareness and interest. We intend to drive patient awareness and interest in part through multiple efforts that may vary over time and may include digital, offline and social marketing. We estimate that there were more than 49 million views of our digital advertisements and more than 6 million views of our digital videos in 2018. We also estimate that visits to our website were 1.7 million in 2018 and searches of our website for physicians capable of placing our Obalon Balloon System were over 580,000 in 2018. We also generated over 71,000 patient leads to our physician partners in the United States during 2018. During the three months ended March 31, 2019, we estimate there were approximately 8.7 million views of our digital advertisements, more than 3.1 million views of our digital videos, over 200,000 visits to our website, over 18,000 searches of our website for physicians capable of placing our Obalon Balloon System and generated approximately 16,000 patient leads to our physician partners in the United States. In the fourth quarter of 2018, we enhanced our capabilities to convert patient interest to treatment by implementing an in-house Ambassador Center with the capabilities to convert the patient interest we generated through our marketed efforts into booked appointments on physicians’ calendars. We believe our Obalon-branded retail center will be able to directly leverage these capabilities and allow us to enhance the patient experience, from initial interests through to the end of patient therapy. |
5
• | Optimize manufacturing to drive operating leverage. We have built a highly leverageable manufacturing facility at our headquarters in Carlsbad, California, where we design, develop and manufacture a majority of our products in-house using some components and sub-assemblies provided by third-party suppliers. We believe that controlling the manufacturing and assembly of our products allows us to innovate more quickly and cost-efficiently and produce higher quality products than if we outsourced manufacturing. We believe we have the ability to increase our manufacturing scale for our current products within our current facility in a cost-effective manner. |
• | Protect and expand our strong intellectual property portfolio. We have developed a strong portfolio of issued patents and pending applications that protect our products and technology. We believe we have also developed know-how critical to creating current and future products that we hold and protect as trade secrets. We have an inventive culture and expect to continue innovating to create a proprietary pathway for future product development. We intend to aggressively protect and enforce our intellectual property, both for existing and new products. |
• | Continue to develop innovative products to facilitate market penetration. We plan to leverage our proprietary product technology and research and development expertise to develop products for weight loss that improve clinical outcomes, increase ease of use and reduce cost. In 2018, we received approvals of PMA-supplements, or PMA-S, for both our Obalon Navigation System and Obalon Touch Inflation Dispenser, which are designed to make balloon placements more reliable, easier, safer and less expensive. Other product candidates currently in our development pipeline include a balloon with a treatment period of longer than six months. |
Our Products and Technology
The Obalon Balloon System was designed to overcome the historical limitations of traditional liquid-filled intragastric balloons and other nonsurgical treatments for weight loss. We have developed the individual components of the Obalon Balloon System to collectively improve clinical outcomes, increase ease of use and reduce cost.
The Obalon Balloon System
The current generation of our Obalon Balloon System consists of a swallowable capsule that contains an inflatable balloon attached to a microcatheter; the Navigation System console, which is a combination of hardware and software used to track and display the location of the balloon during placement; the Obalon Touch Inflation Dispenser, which is a semi-automated, hand-held inflation device used to inflate the balloon once it is placed; and a disposable canister filled with our proprietary mixture of gas.
Capsule, Balloon and Microcatheter Technology
Dissolvable Capsule
We designed the capsule to be large enough to accommodate the folded balloon, yet small enough to be swallowed. The capsule is titrated to optimize dissolution timing. If the capsule dissolves too quickly, the balloon could be prematurely released
6
before entering the stomach, and if too slowly, the patient and physician are inconvenienced by having to wait longer to inflate the balloon.
Balloon Film
Our film is a coextruded, multilayer polymer consisting primarily of nylon and polyethylene. We designed the film to be thin enough to fit into a swallowable capsule, yet stable enough to withstand the chemical and mechanical forces in the stomach. Our film is biocompatible, cost-effective to manufacture, puncture and abrasion resistant, smooth and atraumatic to the stomach’s lining and able to appropriately retain gas.
Balloon Valve
Our balloon valve is an innovative combination of materials, including silicone and titanium, designed to be highly reliable. The valve is small enough to fit into a swallowable capsule and radiopaqued for visibility under digital imaging. A key feature of our valve is the ability to effectively reseal after the inflation catheter is removed to prevent leaks.
Microcatheter
Our microcatheter is designed to quickly and reliably inflate the Obalon balloon. It is small, flexible and smooth in order to minimize any potential discomfort to the patient during balloon placement. The catheter utilizes a hydrophilic coating to reduce friction during swallowing.
Inflation Systems
EzFill Inflation System
Our prior generation hand-held inflation system, the EzFill inflation system, is a reusable device that delivers our proprietary mixture of gas to consistently inflate the Obalon balloon to the standardized volume and pressure. The inflation system is equipped with pre-pulse, a confirmation system that provides pressure feedback measurements to confirm that the Obalon balloon is both properly placed and able to be correctly inflated in the stomach. The EzFill Inflation System is used only with our prior generation Obalon balloon system.
The Obalon Touch Inflation Dispenser
Our new Obalon Touch Inflation Dispenser is a semi-automated, hand-held inflation device that provides real-time balloon pressure measurements to confirm that the Obalon balloon is both properly placed and correctly inflated in the stomach. The Obalon Touch Inflation Dispenser automates several steps of the balloon inflation process and eliminates the need for altitude pre-programming. The Obalon Navigation System is intended to be commercially launched exclusively with the Obalon Touch Inflation Dispenser.
Proprietary Gas
The Obalon balloon is inflated with our proprietary mix of gas, which, in combination with the permeability of the balloon film and the stomach gases, enables the balloon to remain inflated for the full six-month treatment period.
The Obalon Navigation System
The Obalon Navigation System consists of a Navigation console and the Obalon Touch Inflation Dispenser. The Obalon Navigation System console is a portable device consisting of hardware and software that are used to track and display the Navigation balloon during administration. The console has a significantly smaller footprint than most x-ray systems currently used by physicians when placing balloons and does not require any special facilities or licensing. The Obalon Navigation balloon is placed utilizing the Obalon Navigation System console and Obalon Touch Inflation Dispenser. The balloon administered with the Obalon Navigation System is similar to the current balloon but utilizes a new catheter, which interfaces
7
with the Obalon Navigation System console to dynamically track the balloon during placement. The Obalon Navigation Balloon is only compatible with the Obalon Navigation Console and Obalon Touch Inflation Dispenser.
The Obalon Balloon Treatment
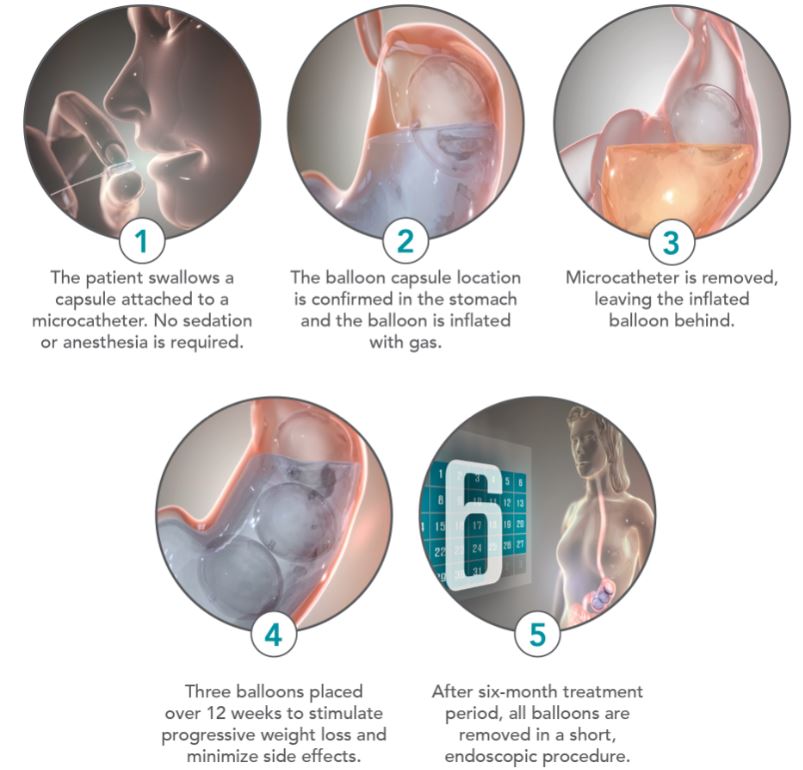
Placement of the Obalon balloon typically occurs in less than 10 minutes and can be accomplished in an outpatient setting. To place the Obalon balloon, the patient swallows the capsule, which has the Obalon balloon folded inside, with a glass of water. No sedation or anesthesia is required. Once swallowed, placement of the capsule is confirmed in the stomach using the Obalon Navigation System. Balloon placement can also be confirmed using x-ray. The microcatheter, which is attached to the Obalon balloon, is then connected to the Obalon Touch Inflation Dispenser. The Touch inflation systems provides real-time pressure measurements to confirm that the Obalon balloon is both properly placed and able to be correctly inflated in the stomach. A pre-filled canister of gas is inserted into the inflation system and then the gas is discharged to fill the balloon to a volume of 250cc. Once the inflation of the Obalon balloon is confirmed, the microcatheter is detached from the balloon via hydrostatic pressure and is removed through the patient’s mouth. The patient is intended to return two more times over the following eight to 12 weeks to receive a second and third Obalon balloon, expanding total balloon volume within the stomach to approximately 750cc.
All of the balloons are removed in a single procedure no more than six months after the placement of the initial balloon. The balloons are removed endoscopically under light conscious sedation, using standard commercially-available endoscopy tools. The endoscopic procedure typically requires approximately 15 minutes on average.
The following pictures depict the treatment steps of the Obalon Balloon System:
8
Additional Product Under Development
We are developing a balloon intended for a longer duration of treatment, potentially up to one year. In our SMART trial, patients in the Obalon treatment group continued, on average, to lose weight throughout the six months of balloon treatment. We have completed the initial engineering and animal testing on the proprietary materials and systems, which we believe would permit reliable balloon performance over a longer period of up to twelve months. We intend to study if longer balloon treatment is safe and may provide greater weight loss in higher BMI patients or those desiring a longer weight loss treatment.
Research and Development
As of June 14, 2019, we had eight employees focused on research and development. In addition to our internal team, we retain third-party contractors from time to time to provide us with assistance on specialized projects. We also work closely with experts in the medical community to supplement our internal research and development resources. Research and development expenses for the years ended December 31, 2018 and 2017 were $10.7 million and $10.6 million, respectively. Research and development expenses for the three months ended March 31, 2019 and 2018 were $2.4 million and $2.6 million, respectively.
Clinical Trials and Data
SMART Trial
9
Based on our clinical data, we believe our Obalon balloon has the potential to offer a compelling combination of efficacy and safety. We have evaluated various versions of our Obalon Balloon System in numerous clinical trials, which included a total of 889 patients as of December 31, 2018. Based on the results of our U.S. pivotal trial, the SMART trial, we received FDA approval for our current Obalon Balloon System in September 2016. The data was published in Surgery for Obesity and Related Diseases in September 2018. The SMART trial met its primary weight loss endpoints, demonstrated a strong safety profile, continued weight loss over the full six-month treatment period, showed statistically significant differences in metabolic profiles and demonstrated that patients were able to maintain most of the weight loss for at least six months following the removal of the Obalon balloons.
The SMART trial was a prospective, double-blinded, multi-center, randomized (1:1), parallel-group, active sham-controlled trial of 387 patients. The Obalon treatment group received three balloons placed individually at approximately week zero, week three and week 12. Alternatively, the sham-control group received placebo capsules with microcatheters and were led to believe in a mock placement that a balloon was placed and inflated in their stomachs at week zero, week three and week 12. Patients were given minimal diet counseling of 25 minutes every three weeks in order to isolate the impact of the Obalon balloon on weight reduction.
The trial was conducted by both bariatric surgeons and gastroenterologists at 15 U.S. centers. The trial evaluated a co-primary endpoint comprised of (i) a minimum difference in mean percent TBL between the Obalon treatment group and sham-control group of at least 2.1% and (ii) achievement by at least 35% of the Obalon treatment group patients of at least 5% TBL at the end of six-months of treatment. Additional observational measures included metabolic metrics and weight loss maintenance after removal of balloons. The median time for each balloon placement was nine minutes, while the median balloon removal time for three balloons was 14 minutes.
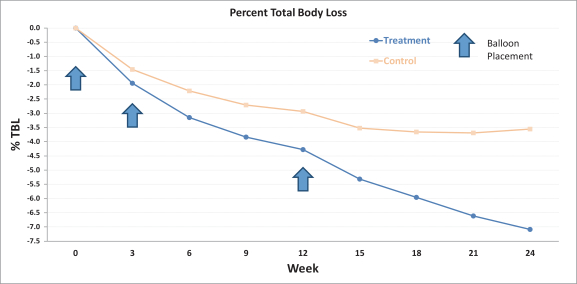
Results from the SMART trial met both the co-primary endpoints. The per protocol analysis included 366 patients (185 in the Obalon treatment group and 181 in the sham-control group) and showed patients in the Obalon treatment group achieved mean TBL of 6.86%, or 15.06 lbs, vs 3.59%, or 7.77 lbs, in the sham-control group, showing a difference of 3.28%, or 7.28 lbs. The following table summarizes average percentage of TBL, percentage of excess weight loss, or EWL, and weight loss (in pounds) for the Obalon treatment group and the sham-control group in the SMART trial. All weight loss metrics below were statistically significant.
Weight Loss Metric Per Protocol Cohort | Obalon Treatment Group (N = 185) | Sham-Control Group (N = 181) | Difference | p-value |
Percent TBL | -6.86 | -3.59 | -3.28 | 0.0261 |
Percent EWL | -25.05 | -12.95 | -12.09 | < 0.0001 |
Weight Loss (lbs.) | -15.06 | -7.77 | -7.28 | < 0.0001 |
In addition, 64.9% of the Obalon treatment group patients met or exceeded the 5% TBL endpoint whereas only 32.0% of the sham-control group met or exceeded 5% TBL. The following table summarizes the 5% TBL responder rates for the Obalon treatment group and the sham-control group in the SMART trial.
Main Analysis of -5% TBL Responder Rate | Estimate |
Obalon Treatment Group—Per Protocol Cohort* | 120 / 185 (64.9%) |
Sham-Control Group | 58 / 181 (32.0%) |
Difference (Treatment less Control) | 32.8% |
* | p-value <0.0001 |
10
The following table summarizes the various responder rate thresholds for the Obalon treatment group and the sham-control group in the SMART trial.
Responder Rate Threshold (-%TBL) | Obalon Treatment Group | Sham-Control Group |
-6% | 98 / 185 (53.0%) | 47 / 181 (26.0%) |
-7% | 81 / 185 (43.8%) | 38 / 181 (21.0%) |
-8% | 68 / 185 (36.8%) | 35 / 181 (19.3%) |
-9% | 55 / 185 (29.7%) | 29 / 181 (16.0%) |
-10% | 49 / 185 (26.5%) | 23 / 181 (12.7%) |
Notably, the Obalon treatment group demonstrated a progressive weight loss profile for the duration of the six-month therapy period. The following chart shows percent TBL by week for the Obalon treatment group and sham-control group. The arrows represent the average week of each balloon placement.
In addition, nearly all patients in the Obalon treatment group, including patients in the bottom 25% of the group, achieved TBL, EWL and weight loss and a reduction in BMI. The table below summarizes the mean, the average of the top 25% of the results, the average of the bottom 25% of the results and the single best changes in TBL, EWL, weight loss and BMI achieved by patients in the Obalon treatment group.
Weight Loss Metric | Mean | Average Top 25% | Average Worst 25% | Single Best |
Percent TBL | -6.9% | -10.2% | -3.6% | -19.3% |
Percent EWL | -25.1% | -36.3% | -12.3% | -80.7% |
Weight Loss (lbs.) | -15.1 | -21.8 | -7.4 | -49.7 |
BMI Change | -2.4 | -3.6 | -1.3 | -7.1 |
In an observational analysis at six months, the Obalon treatment group also demonstrated statistically significant improvements in systolic blood pressure, fasting glucose, total cholesterol and triglycerides compared to both their own baseline measures and to the sham-control group.
11
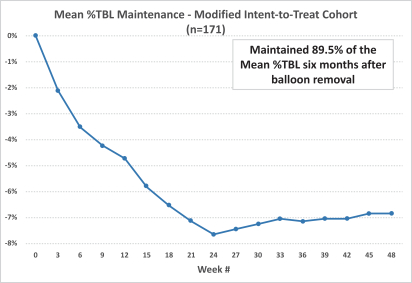
At the conclusion of the six-month treatment period, the Obalon treatment group patients continued with the standardized behavior modification program for six additional months after the Obalon balloon removal. An additional observational data analysis of the subjects who lost weight in the first six months of the study and were evaluated for up to an additional six months, suggests that, on average, 89.5% of the weight loss was maintained six months after balloon removal. The following graph depicts the weight loss maintained for the one-year period in the Obalon treatment group. We did not continue to collect data from patients in the sham-control group who received the Obalon balloons subsequent to balloon removal.
As part of the SMART trial, we actively solicited patients to provide details of any adverse events, or AEs, by contacting all patients 24 hours after each Obalon balloon placement and balloon removal as well as at every office visit. All AEs were first assigned a device-relatedness and a pre-defined severity rating. Mild events did not require intervention, required homeopathic remedies (including chamomile tea, peppermint oil tea and Altoids) or required over the counter remedies to treat and resolve the events. Moderate severity events required a prescription medication to treat and resolve the event. Severe events required medical intervention beyond a prescription medication.
In our SMART trial, only one out of 336 patients (0.3%) receiving Obalon balloons in both phases experienced a SADE. The event was described as peptic ulcer disease, or bleeding. The patient was hospitalized, and after stabilization, the patient was discharged from the hospital without sequelae. During the Obalon balloon therapy period the subject underwent an outpatient total knee replacement surgery. During the surgery and as part of post-operative recovery, the subject was prescribed both a high dose of nonsteroidal anti-inflammatory drugs, or NSAIDs, and aspirin, both of which are contraindicated for use with each other as well as for use in conjunction with the Obalon Balloon System. The SADE event was determined to be “possibly,” but not “probably,” device-related by the investigator since concomitant high dose NSAID and aspirin use is also known to cause peptic ulcer disease. The investigator felt that the NSAID and aspirin use was the primary cause of the event but could not rule out the balloons completely. The patient previously had no ulcers per the upper gastrointestinal screen performed at time of enrollment and was not taking medications prior to surgery.
In our SMART trial, there were no surgical removals or other hospitalizations due to a SADE other than the SADE described above. The most common other adverse device events during balloon placement were abdominal pain (72.6% of patients), nausea (56.0% of patients) and vomiting (17.3% of patients), all of which were classified as mild or moderate.
12
Commercial-Use Patient Registry
In order to closely monitor the safety, efficacy and quality of the Obalon Balloon System in actual commercial use, we have created an online clinical performance database, or registry. Product credits were provided to physician customers for entering data from new patients starting therapy in the United States from January 2017 through June 2018. All physicians and institutions using the Obalon Balloon System have been encouraged to enter their patient data in the registry and compare their performance to national and regional data. The data collected in the registry includes gender, initial height and weight, weights at each subsequent balloon placement, weight at removal, adverse events occurring during the treatment, and product quality and performance.
Data on the first full year of commercialization of the Obalon Balloon System was recently published in Surgery for Obesity and Related Diseases. Data on demographics, balloon placement timing, weight loss, adverse events, and product performance were prospectively captured in the registry and retrospectively analyzed on 1,387 consecutive patients who initiated treatment in the first year of commercialization at 108 treating sites. A retrospective analysis of 1,343 (97%) patients entered who met the predefined analyses protocol definitions was approved by an Institutional Review Board. This data is self-reported by the physicians or institutions and we do not perform a formal audit of the data. However, the registry was validated and contains embedded edit checks to ensure data accuracy and completeness.
Demographics
Mean baseline demographics presented in the publication were: age 45.7±10.8 years, BMI 35.4±5.4 kg/m2, height 65.9±3.5 inches, weight 219.5± 42.9 lbs., female 78.6% and white 66.8%.
Safety
The manuscript reported no deaths or unanticipated adverse events. Two serious adverse events were reported, corresponding to 0.15% of patients. There were 308 non-serious adverse events reported in 14.2% of the patients. The most frequent adverse events reported were abdominal pain (5.3%), nausea (4.7%), vomiting (2.3%) and abdominal distension (1.0%). The remaining adverse events were less than 1.0%.
Weight Loss
The weight loss reported in the manuscript in patients meeting the product indication (BMI 30-40 kg/m2 with 3 balloons for ≥ 20 weeks of therapy) was 21.3 ± 13.5 lbs., 10.0% ± 6.1% of total body weight loss (TBWL), 38.3% ± 25.3% excess weight loss (EWL) and a 3.4 ± 2.1 reduction in BMI. Of note, the top quartile of those patients lost an average of 38.2 pounds, resulting in a 17.2% reduction in total body weight and a 6.1 point decrease in BMI compared to baseline values. Average weight loss across all patients with a BMI>25 was 21.7 lbs resulting in a percent total body loss of 9.9%. The top quartile of all patients with a BMI>25 lost an average of 39.0 lbs., resulting in a 16.8% reduction in total body weight and a 6.2 point decrease in BMI compared to baseline values. We believe the outcome data collected in this registry is the largest known registry of an approved endoscopic bariatric therapy to date, including intragastric balloons, and provides evidence of effective weight loss and safety in a real-world, commercial setting. The data captured in the registry for the first year of commercialization (January 9, 2017 to December 31, 2017) was accepted for publication in the journal Surgery for Obesity and Related Diseases.
Commercial Safety Experience
As of December 31, 2018, the SADEs reported to us in commercial use remain consistent with the total rate reported in the SMART Study. Since we began selling in United States in January 2017, we have reported adverse events relating to potential or actual patient injuries associated with use of the Obalon balloon in the FDA's MAUDE database.
Post-approval study - Obalon Balloon System
13
To help assure the continued safety and effectiveness of the Obalon Balloon System, the FDA has required a post-approval study as a condition of approval under 21 CFR 814.82(a)(2). As part of our PMA approval, we are required to conduct a post-approval study that will evaluate 200 patients who will be enrolled at a maximum of 15 sites in the United States. The study is a prospective, open-label, single-arm, 12-month follow-up study in which patients will be treated during the first six months with placement of up to three Obalon balloons in conjunction with a moderate intensity weight loss and behavioral modification program standardized throughout the sites, followed by observational evaluation for an additional six months after device removal. The primary endpoint is to evaluate the safety of the Obalon Balloon System by assessing the rate of device- or procedure-related serious adverse events. We are required to submit an Interim Post-Approval Study Status Report every six months after the date of PMA approval for the first two years of the study and annually thereafter until 200 patients have completed the study. We are currently enrolling this study.
Post-approval study - Obalon Navigation System with Touch Dispenser
To help assure the continued safety and effectiveness of the Obalon Navigation System, the FDA has required a post-approval study as a condition of approval under 21 CFR 814.82(a)(2). As part of our PMA approval, we are required to conduct a post-approval study that will evaluate a minimum of 1,000 commercial patients and 3,689 balloon administrations across 40 clinical sites in the United States. The study will be a prospective, observational, open-label, multi-center study designed to capture additional information to demonstrate the continued safety of the administration of Obalon balloons with NTS to collect acute safety and efficacy data surrounding balloon placement; no long-term data, such as weight loss at six-months is required. The study will have a single cohort group that includes subjects who commercially purchased the Obalon Balloon System intended to be administered with NTS and have consented to have their data collected to support this study. All activities related to post-administration management and removal of the balloons will be conducted in accordance with the commercial Obalon Balloon System device labeling and will not be collected in this study; this study will focus on balloon administrations only.
Sales and Marketing
Our primary selling efforts are conducted in the United States, with some sales historically generated through distributors in select international markets. In the United States, we sell our product to physicians through a centralized customer support model that includes marketing and clinical support from our corporate office. Marketing support may include media assets and media purchasing support, access to our call center, leads generated from our Find-A-Doc locater and leads generated from our digital and off-line marketing efforts. Clinical support may include on-site and remote physician and staff training. We believe our centralized customer support model encompasses the three key disciplines that we believe are necessary to develop the market for our Obalon Balloon System in the United States: sales conversion, practice development and clinical training and application. We also intend to establish a company-owned or managed Obalon-branded retail center strategy. Under the Obalon-branded retail strategy, we plan to either directly employ physicians or manage the practices where the treatments are prescribed by physicians, and we would provide the physician office support staff, equipment and services necessary for patients to receive the Obalon balloon therapy. We believe this model will contribute to standardization of both quality of care and patient pricing and provide us greater operational and financial control of our business. We plan to launch our first Obalon-branded retail center before year end 2019. We are not currently selling in international markets, but plan to utilize distributors should we so chose.
Our U.S. marketing efforts have focused on differentiating the benefits of our technology, leveraging the strong clinical outcome from our SMART trial and peer-review published commercial registry data, working with key physicians in bariatrics, gastroenterology, and plastic surgery, and partnering with physicians to create consumer awareness and drive patients into the channel. We also have provided physicians with the clinical training to utilize our Obalon Balloon System, as well as the practice development support to manage their practices as self-pay centers.
We also intend to continue to drive consumer awareness and interest in part through multiple efforts that may vary over time and may include digital, offline and social marketing. We estimate that there were more than 49 million views of our digital advertisements and more than 6 million views of our digital videos in 2018. We also estimate that visits to our website were 1.7
14
million in 2018 and searches of our website for physicians capable of placing our Obalon Balloon System were over 580,000 in 2018. We also generated over 71,000 patient leads to our physician partners in the United States during 2018. During the three months ended March 31, 2019, we estimate there were approximately 8.7 million views of our digital advertisements, more than 3.1 million views of our digital videos, over 200,000 visits to our website, over 18,000 searches of our website for physicians capable of placing our Obalon Balloon System and generated approximately 16,000 patient leads to our physician partners in the United States.
In late 2018 we developed the internal capabilities and staffing and transitioned from an outside third party to in-house support of a call center, which we have named the Obalon Ambassador Center. We believe our in-house call center could potentially reduce patient acquisition costs and improve the patient experience through a standardized approach from initial awareness up to and including a booked appointment with the physician. We also believe these capabilities may be utilized to support the company-owned or managed Obalon-branded retail center strategy, offering the full range of the patient experience starting with patient awareness through the entire treatment journey. Historically we have sold our products directly to physicians, who would then sell weight loss treatment packages to their patients that included our balloon therapy, dietary counseling and balloon removal on a non-reimbursed, self-pay basis. We are currently undergoing a process to fundamentally change our commercialization efforts, which commenced with the elimination of our direct sales force in connection with an overall workforce reduction in April 2019. Following this transition, in addition to selling directly to physicians under a new centralized customer support model, we intend to establish company-owned or managed Obalon branded retail centers. Under the Obalon branded retail center model, subject to state law requirements, we plan to either directly employ physicians or manage the practices where the treatments are prescribed by physicians, and we will be directly responsible for practice marketing and promotion efforts. We would provide the physician office support staff, equipment and services necessary for patients to receive the Obalon balloon therapy prescribed by licensed physicians. We believe this model will contribute to standardization of both quality of care and patient pricing and provide us greater operational and financial control of our business. We plan to launch our first Obalon branded retail center before year end 2019.
We have limited experience as a company in the sales and marketing of our products or management of operations for treatment of patients. Identifying and recruiting qualified employees and training them in the use of our Obalon Balloon System to achieve the level of clinical competency expected by physicians, and compliance with applicable federal and state laws and regulations and our internal policies and procedures, requires significant time, expense and attention. It can take several months before our employees are fully trained and productive.
Manufacturing
All of our products except the Obalon Navigation System console are manufactured or assembled in-house using components and sub-assemblies at our single-site facility in Carlsbad, California. We rely on single suppliers for the extruded film, swallowable capsule, molded silicone valve used to manufacture our Obalon balloons, the hydrophilic coating for our catheters, the Obalon Navigation System console components and the sensors utilized in the Obalon Navigation balloon catheter. There are minimum purchase requirements and delivery requirements with the supplier for the Obalon Navigation System console and sensor utilized in the Obalon Navigation balloon catheter. Our suppliers for all other components of the Obalon balloon have no contractual obligations to supply us with, and we are not contractually obligated to purchase any of our supplies from them. Order quantities and lead times for components purchased from our suppliers are based on our forecasts derived from historical demand and anticipated future demand. In June 2019 we initiated a facility reduction plan which included consolidating certain functions of manufacturing. There are certain testing and regulatory requirements we must satisfy to restart our manufacturing operations. We expect to satisfy regulatory requirements and resume manufacturing operations in the third quarter 2019. There is no assurance we will be able to satisfy regulatory requirements in a timely manner or at all.
Lead times for components may vary significantly depending on the size of the order, time required to fabricate and test the components, specific supplier requirements and current market demand for the components and subassemblies. These components are critical to our products and there are relatively few alternative sources of supply. Inventory of these components is dependent on several factors including lead times, forecast accuracy, and actual demand, which can lead to more
15
or less inventory than required. Identifying and qualifying additional or replacement suppliers for any of the components or sub-assemblies used in our products could involve significant time and cost, and may delay our commercialization efforts.
We have registered with the FDA as a medical device manufacturer and have obtained a manufacturing license from the Center for Devices and Radiological Health. We and our component suppliers are required to manufacture our products in compliance with the FDA’s Quality System Regulation, or QSR, codified in 21 CFR part 820. The QSR regulates extensively the methods and documentation of the design, testing, control, manufacturing, labeling, quality assurance, packaging, storage and shipping of our products. The FDA enforces the QSR through periodic inspections that may include the manufacturing facilities of our subcontractors. Our quality system has undergone periodic FDA audits, the last of which occurred in November 2017, which resulted in no observations.
Although we expect our third-party suppliers to supply us with components that meet our specifications and comply with regulatory and quality requirements, we do not control our suppliers outside of our agreements, as they operate and oversee their own businesses. There is a risk that our suppliers will not always act consistent with our best interests, and may not always supply components that meet our needs. This risk may be increased as with any new product launch, there is increased risk for supply shortages or product quality issues. Any significant delay or interruption in the supply of components or sub-assemblies, or our inability to obtain substitute components, sub-assemblies or materials from alternate sources at acceptable prices in a timely manner, could impair our ability to meet the demand of our customers and harm our business. We have experienced and may continue to experience production challenges due to shortages of key components from suppliers.
Additionally, we may need to increase our manufacturing capabilities in order to satisfy expected demand for our Obalon Balloon System, and we have no experience manufacturing our Obalon Balloon System in such quantities. If we are unable to keep up with demand for our Obalon Balloon System, our revenue could be impaired, market acceptance for our Obalon Balloon System could be harmed and our customers might instead purchase our competitors’ products.
Competition
The medical device industry generally, and the market for weight loss devices specifically, are highly competitive, subject to rapid change and significantly affected by new product introductions, results of clinical research, corporate combinations, actions by regulatory bodies, changes by public and private payers and other factors. Because of the market opportunity and the high growth potential of the non-surgical device market for weight loss and obesity, competitors and potential competitors have historically dedicated, and will continue to dedicate, significant resources to aggressively develop and commercialize their products.
In the United States, our product competes with a variety of pharmaceuticals, surgical procedures and devices for the treatment of obese and overweight people. There are several competitors in the pharmaceutical segment including Vivus, Inc., Eisai Co., Ltd, Inc., AstraZeneca plc, and Allergan plc. Large competitors in the surgical segment for weight loss and obesity include Ethicon Inc. (subsidiary of Johnson & Johnson), Medtronic plc (formerly Covidien Ltd.), Apollo EndoSurgery, Inc., and ReShape LifeSciences (which acquired the Lap-Band from Apollo Endosurgery, Inc. and currently sells that device worldwide). In addition, we are aware of at least two FDA approved liquid-filled balloon devices for treating overweight people, including the ReShape Duo Balloon and the ORBERA Balloon, both of which are now owned by Apollo EndoSurgery. Outside of the United States, Allurion Technologies, Inc. has developed a swallowable, passable liquid-filled intragastric balloon that has been approved for sale in Europe and the Middle East and completed enrollment in a U.S. clinical trial; and Spatz Medical has also developed a liquid-filled intragastric balloon that has been approved for sale in Latin America and Europe and is currently engaged in a U.S. clinical trial. We also compete against ReShape LifeSciences’ Maestro device, which is intended to create weight loss by vagal nerve stimulation and Aspire Bariatrics' ApireAssist device. Gelesis has developed a hydrogel technology that is intended to expand in the stomach by absorbing water to create the feeling of satiety and its Plentity device was recently cleared by FDA. BAROnova recently gained FDA approval for its transpyloric shuttle, a non-surgical, non-pharmacologic device to induce weight loss by slowing gastric emptying. Additionally, we are aware of numerous companies around the world
16
working to develop less invasive and less costly alternatives for the treatment of obesity, any of which, if approved, could compete with us in the future.
At any time, these or other competitors may introduce new or alternative products that compete directly or indirectly with our products and services. They may also develop and patent products and processes earlier than we can or obtain regulatory clearance or approvals faster than us, which could impair our ability to develop and commercialize similar products or services. If clinical outcomes of procedures performed with our competitors’ products are, or are perceived to be, superior to treatments performed with our products, sales of our products could be negatively affected and our business, results of operations and financial condition could suffer.
Many of our competitors have significantly greater financial and other resources than we do, as well as:
• | well-established reputations and name recognition with key opinion leaders and physician networks; |
• | an established base of long-time customers with strong brand loyalty; |
• | products supported by long-term data; |
• | longer operating histories; |
• | significantly larger installed bases of equipment; |
• | greater existing market share in the obesity and weight management market; |
• | broader product offerings and established distribution channels; |
• | greater ability to cross-sell products; |
• | additional lines of products, and the ability to offer rebates or bundle products to offer higher discounts or incentives; and |
• | more experience in conducting research and development, manufacturing, performing clinical trials and obtaining regulatory approvals or clearances. |
Competition with these companies could result in significant price-cutting, reduced profit margins and loss of market share, any of which would harm our business, financial condition and results of operations. In addition, competitors with greater financial resources than ours could acquire other companies to gain enhanced name recognition and market share, as well as new technologies or products that could effectively compete with our existing and future products, which may cause our revenues to decline and harm our business.
In order to compete effectively, we plan to continue to develop new product offerings and enhancements to our existing Obalon Balloon System, price our product competitively with traditional liquid-filled intragastric balloons and maintain adequate research and development and sales and marketing personnel and resources to meet the demands of the market.
Intellectual Property
In order to remain competitive, we must develop and maintain protection of the proprietary aspects of our technologies. We rely on a combination of patents, trademarks, trade secret laws and confidentiality and invention assignment agreements to protect our intellectual property rights.
It is our policy to require our employees, consultants, contractors, outside scientific collaborators and other advisers to execute non-disclosure and assignment of invention agreements on commencement of their employment or engagement. Agreements with our employees also forbid them from using the proprietary rights of third parties in their work for us. We also require third parties that receive our confidential data or material to enter into confidentiality or material transfer agreements.
17
As of June 14, 2019, we held 20 issued U.S. patents and had 26 pending U.S. patent applications, as well as 31 international patents issued in regions including Europe, Mexico, Australia, Canada, Asia, China and Israel and 54 pending international patent applications in regions including Australia, Canada, Europe, Asia, the Middle East and South America. Our issued patents expire between the years 2023 and 2036, and are directed to various features and combinations of features of the Obalon Balloon System technology, including the apparatus for connecting the balloon to an inflation catheter, the structure and composition of the balloon wall, and the composition of the initial fill gas.
Our patent applications may not result in issued patents and our patents may not be sufficiently broad to protect our technology. Any patents issued to us may be challenged by third parties as being invalid or unenforceable, or third parties may independently develop similar or competing technology that does not infringe our patents. The laws of certain foreign countries do not protect our intellectual property rights to the same extent as do the laws of the United States.
As of June 14, 2019, we held two registered U.S. trademarks and 35 registered marks throughout Europe, the Middle East, Asia and Mexico. We have five pending U.S. trademark applications and 6 pending marks outside the United States, including in Europe, the Middle East, Asia and Mexico.
Geographic Regions
Substantially all of our assets, and the majority of revenues and expenses for 2018 and 2017 were located in or derived from operations in the United States. In addition, we have had sales through Bader in the Middle East. During 2018 and 2017, international revenues accounted for approximately 48.4% and 16.7%, respectively, of our total revenues. International revenues represented 33.1% and 63.1% of our total revenue for the three months ended March 31, 2019 and 2018, respectively. In the first quarter of 2019, we completed final shipments of the current generation product to Bader, and going forward we intend to focus our selling efforts on the United States. As a result, we do not anticipate additional international revenue in 2019.
Seasonality
We have limited experience selling our product in the United States and have realized significant volatility in quarterly balloon sales. As a result, we are unable to discern seasonal variations in demand for our products. In the future, seasonal fluctuations in the number of patients seeking treatment and the availability of our customers may affect our business.
Government Regulation
Our products and operations are subject to extensive and rigorous regulation by the FDA and other federal, state and local authorities, as well as foreign regulatory authorities. The FDA regulates, among other things, the research, development, testing, design, manufacturing, approval, labeling, storage, recordkeeping, advertising, promotion and marketing, distribution, post approval monitoring and reporting and import and export of medical devices (such as the Obalon Balloon System) in the United States to assure the safety and effectiveness of medical products for their intended use. The Federal Trade Commission also regulates the advertising of our products in the United States. Further, we are subject to laws directed at preventing fraud and abuse, which subject our sales and marketing, training and other practices to government scrutiny.
Regulatory System for Medical Devices in the United States
Unless an exemption applies, each new or significantly modified medical device we seek to commercially distribute in the United States will require either a premarket notification to the FDA requesting permission for commercial distribution under Section 510(k) of the Federal Food, Drug and Cosmetic Act, or FFDCA, also referred to as a 510(k) clearance, or approval from the FDA of a PMA application. Both the 510(k) clearance and PMA processes can be resource intensive, expensive, and lengthy, and require payment of significant user fees, unless an exemption is available.
18
Device Classification
Under the FFDCA, medical devices are classified into one of three classes—Class I, Class II or Class III—depending on the degree of risk associated with each medical device and the extent of control needed to provide reasonable assurances with respect to safety and effectiveness.
Class I includes devices with the lowest risk to the patient and are those for which safety and effectiveness can be reasonably assured by adherence to a set of FDA regulations, referred to as the General Controls for Medical Devices, which require compliance with the applicable portions of the QSR, facility registration and product listing, reporting of adverse events and malfunctions, and appropriate, truthful and non-misleading labeling and promotional materials. Some Class I devices, also called Class I reserved devices, also require premarket clearance by the FDA through the 510(k) premarket notification process described below. Most Class I products are exempt from the premarket notification requirements.
Class II devices are those that are subject to the General Controls, and special controls as deemed necessary by the FDA to ensure the safety and effectiveness of the device. These special controls can include performance standards, patient registries, FDA guidance documents and post-market surveillance. Most Class II devices are subject to premarket review and clearance by the FDA. Premarket review and clearance by the FDA for Class II devices is accomplished through the 510(k) premarket notification process.
Class III devices include devices deemed by the FDA to pose the greatest risk such as life-supporting or life-sustaining devices, or implantable devices, in addition to those deemed novel and not substantially equivalent following the 510(k) process. The safety and effectiveness of Class III devices cannot be reasonably assured solely by the General Controls and Special Controls described above. Therefore, these devices are subject to the PMA application process, which is generally more costly and time consuming than the 510(k) process. Through the PMA application process, the applicant must submit data and information demonstrating reasonable assurance of the safety and effectiveness of the device for its intended use to the FDA’s satisfaction. Accordingly, a PMA application typically includes, but is not limited to, extensive technical information regarding device design and development, pre-clinical and clinical trial data, manufacturing information, labeling and financial disclosure information for the clinical investigators in device studies. The PMA application must provide valid scientific evidence that demonstrates to the FDA’s satisfaction a reasonable assurance of the safety and effectiveness of the device for its intended use.
The Investigational Device Process
In the United States, absent certain limited exceptions, human clinical trials intended to support medical device clearance or approval require an IDE application. Some types of studies deemed to present “non-significant risk” are deemed to have an approved IDE once certain requirements are addressed and IRB approval is obtained. If the device presents a “significant risk” to human health, as defined by the FDA, the sponsor must submit an IDE application to the FDA and obtain IDE approval prior to commencing the human clinical trials. The IDE application must be supported by appropriate data, such as animal and laboratory testing results, showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. The IDE application must be approved in advance by the FDA for a specified number of subjects. Generally, clinical trials for a significant risk device may begin once the IDE application is approved by the FDA and the study protocol and informed consent are approved by appropriate institutional review boards at the clinical trial sites. There can be no assurance that submission of an IDE will result in the ability to commence clinical trials, and although the FDA’s approval of an IDE allows clinical testing to go forward for a specified number of subjects, it does not bind the FDA to accept the results of the trial as sufficient to prove the product’s safety and efficacy, even if the trial meets its intended success criteria.
All clinical trials must be conducted in accordance with the FDA’s IDE regulations that govern investigational device labeling, prohibit promotion and specify an array of recordkeeping, reporting and monitoring responsibilities of study sponsors and study investigators. Clinical trials must further comply with the FDA’s good clinical practice regulations for institutional review board approval and for informed consent and other human subject protections. Required records and reports are subject to
19
inspection by the FDA. The results of clinical testing may be unfavorable, or, even if the intended safety and efficacy success criteria are achieved, may not be considered sufficient for the FDA to grant marketing approval or clearance of a product.
The PMA Approval Process
Following receipt of a PMA application, the FDA conducts an administrative review to determine whether the application is sufficiently complete to permit a substantive review. If it is not, the agency will refuse to file the PMA. If it is, the FDA will accept the application for filing and begin the review. The FDA, by statute and by regulation, has 180 days to review a filed PMA application, although the review of an application more often occurs over a significantly longer period of time. During this review period, the FDA may request additional information or clarification of information already provided, and the FDA may issue a major deficiency letter to the applicant, requesting the applicant’s response to deficiencies communicated by the FDA. The FDA considers a PMA or PMA supplement to have been voluntarily withdrawn if an applicant fails to respond to an FDA request for information (e.g., major deficiency letter) within a total of 360 days. Before approving or denying a PMA, an FDA advisory committee may review the PMA at a public meeting and provide the FDA with the committee’s recommendation on whether the FDA should approve the submission, approve it with specific conditions, or not approve it. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Prior to approval of a PMA, the FDA may conduct inspections of the clinical trial data and clinical trial sites, as well as inspections of the manufacturing facility and processes. Overall, the FDA review of a PMA application generally takes between one and three years, but may take significantly longer.
If the FDA evaluation of a PMA is favorable, the FDA will issue either an approval letter, or an approvable letter, the latter of which usually contains a number of conditions that must be met in order to secure final approval of the PMA. When and if those conditions have been fulfilled to the satisfaction of the FDA, the agency will issue a PMA approval letter authorizing commercial marketing of the device, subject to the conditions of approval and the limitations established in the approval letter. If the FDA’s evaluation of a PMA application or manufacturing facilities is not favorable, the FDA will deny approval of the PMA or issue a not approvable letter. The FDA also may determine that additional tests or clinical trials are necessary, in which case the PMA approval may be delayed for several months or years while the trials are conducted and data is submitted in an amendment to the PMA, or the PMA is withdrawn and resubmitted when the data are available. The PMA process can be expensive, uncertain and lengthy and a number of devices for which the FDA approval has been sought by other companies have never been approved by the FDA for marketing.
New PMA applications or PMA supplements are required for modification to the manufacturing process, equipment or facility, quality control procedures, sterilization, packaging, expiration date, labeling, device specifications, ingredients, materials or design of a device that has been approved through the PMA process. PMA supplements often require submission of the same type of information as an initial PMA application, except that the supplement is limited to information needed to support any changes from the device covered by the approved PMA application and may or may not require as extensive technical or clinical data or the convening of an advisory panel, depending on the nature of the proposed change.
In approving a PMA application, as a condition of approval, the FDA may also require some form of post-approval study or post-market surveillance, whereby the applicant conducts a follow-up study or follows certain patient groups for a number of years and makes periodic reports to the FDA on the clinical status of those patients when necessary to protect the public health or to provide additional or longer term safety and effectiveness data for the device. The FDA may also require post-market surveillance for certain devices cleared under a 510(k) notification, such as implants or life-supporting or life-sustaining devices used outside a device user facility. The FDA may also approve a PMA application with other post-approval conditions intended to ensure the safety and effectiveness of the device, such as, among other things, restrictions on labeling, promotion, sale, distribution and use. Intragastric balloons, including the Obalon Balloon System, are considered Class III medical devices. In order to support a PMA application, the FDA required us to conduct a large, rigorous and expensive, double-blinded, randomized, sham-controlled trial. We will be required to file new PMA applications or PMA supplement applications for
20
modifications to our PMA-approved Obalon Balloon System and Obalon Navigation System or any of its components, including modifications to our manufacturing processes, device labeling and device design, based on the findings of post-approval studies.
Pervasive and Continuing FDA Regulation
After the FDA permits a device to enter commercial distribution, numerous regulatory requirements continue to apply. These include:
• | the FDA’s QSR, which requires manufacturers, including third party manufacturers, to follow stringent design, testing, production, control, supplier/contractor selection, complaint handling, documentation and other quality assurance procedures during all aspects of the manufacturing process; |
• | labeling regulations, unique device identification requirements and FDA prohibitions against the promotion of products for uncleared, unapproved or off-label uses; |
• | advertising and promotion requirements; |
• | restrictions on sale, distribution or use of a device; |
• | PMA annual reporting requirements; |
• | PMA approval of product modifications; |
• | medical device reporting, or MDR, regulations, which require that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction were to recur; |
• | medical device correction and removal reporting regulations, which require that manufacturers report to the FDA field corrections and product recalls or removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the FDCA that may present a risk to health; |
• | recall requirements, including a mandatory recall if there is a reasonable probability that the device would cause serious adverse health consequences or death; |
• | an order of repair, replacement or refund; |
• | device tracking requirements; and |
• | post-market surveillance regulations, which apply when necessary to protect the public health or to provide additional safety and effectiveness data for the device. |
In addition, FDA enforces the Medical Device Reporting, or MDR, regulations, which require that we report to the FDA any incident in which our product may have caused or contributed to a death or serious injury or in which our product malfunctioned and, if the malfunction were to recur, would likely cause or contribute to death or serious injury. Since February 2017, the FDA has issued three separate Letters to Health Care Providers warning of serious adverse events, including deaths, which are specific to liquid-filled intragastric balloons. We are aware of the filing of additional reports of serious adverse events, including deaths, associated with liquid-filled balloons since the issuance of the Safety Alerts. While the Safety Alerts were specific to liquid-filled intragastric balloons and not the Obalon gas-filled balloons, these letters could create negative perceptions of the entire gastric balloon category which may cause negative consequences for us including requiring additional warnings, precautions and/or contraindications in the labeling than originally required, delaying or denying approval of our
21
future products, or possible review or withdrawal of our current approval. Since we began selling in United States in January 2017, we have reported adverse events relating to patient injuries associated with use of the Obalon balloon in the FDA's MAUDE database.
The FDA has broad post-market and regulatory enforcement powers. Medical device manufacturers are subject to unannounced inspections by the FDA and other state, local and foreign regulatory authorities to assess compliance with the QSR and other applicable regulations, and these inspections may include the manufacturing facilities of any suppliers.
Failure to comply with applicable regulatory requirements can result in enforcement action by the FDA, which may include any of the following sanctions:
• | warning letters, untitled letters, Form 483s, fines, injunctions, consent decrees and civil penalties; |
• | unanticipated expenditures, repair, replacement, refunds, recall or seizure of our products; |
• | operating restrictions, partial suspension or total shutdown of production; |
• | the FDA’s refusal of our requests for 510(k) clearance or premarket approval of new products, new intended uses or modifications to existing products; |
• | the FDA’s refusal to issue certificates to foreign governments needed to export products for sale in other countries; |
• | withdrawing 510(k) clearance or premarket approvals that have already been granted; and |
• | criminal prosecution. |
Regulatory System for Medical Devices in Europe
The European Union consists of member states residing in the European Union and has a coordinated system for the authorization of medical devices. The European Union Medical Devices Directive, or MDD, sets out the basic regulatory framework for medical devices in the European Union. This directive has been separately enacted in more detail in the national legislation of the individual member states of the European Union.
The system of regulating medical devices operates by way of a certification for each medical device. Each certificated device is marked with CE mark which shows that the device has a Certificat de Conformité. There are national bodies known as Competent Authorities in each member state which oversee the implementation of the MDD within their jurisdiction. The means for achieving the requirements for CE mark varies according to the nature of the device. Devices are classified in accordance with their perceived risks, similarly to the U.S. system. The class of a product determines the requirements to be fulfilled before CE mark can be placed on a product, known as a conformity assessment. Conformity assessments for our products are carried out as required by the MDD. Each member state can appoint Notified Bodies within its jurisdiction. If a Notified Body of one member state has issued a Certificat de Conformité, the device can be sold throughout the European Union without further conformance tests being required in other member states.
According to the MDD, the Obalon Balloon System, when delivered with a porcine capsule, is considered a Class III product. We discontinued sales of the Obalon Balloon System with a porcine capsule in 2017. The Obalon Balloon System when delivered with a cellulose-based capsule is considered a Class IIb product. We believe the Obalon Navigation System and the Obalon Touch Inflation Dispenser are Class I products and will follow a similar pathway as the EzFill Dispenser. We have not applied for a CE-mark for the Obalon Navigation System and Obalon Touch Inflation Dispenser at this time.
Regulatory Frameworks for Medical Devices in Certain Countries in the Middle East
Unlike Europe, while the Gulf Cooperation Council, or GCC, jurisdictions often work together to purchase certain medical products in a coordinated fashion for government hospitals, there is not a coordinated system for the authorization of medical
22
devices. Most GCC jurisdictions require that the official registered distributor of a product be wholly owned by nationals of that particular GCC jurisdiction.
Kingdom of Saudi Arabia, or KSA
The most pertinent regulation is the Interim Regulation for Medical Devices, issued by the Saudi Food & Drug Authority, or SFDA, Board of Directors’ Decree number 1-8-1429 dated approximately December 27, 2008 and the implementing regulations of the same. The SFDA is an independent regulatory body that is responsible for the authorization of medical devices, and current guidelines are generally based on pre-existing approval in one of the five founding member nations of the Global Harmonization Task Force, or GHTF, which are Australia, Canada, United States, European Union and Japan. There are no overt requirements for the provision of safety and effectiveness data in the form of clinical trials or other studies but these would likely come as a part of the approvals described above that are used as a basis to support approval within the KSA. The SFDA reserves its rights to require its own independent clinical trials as it deems necessary or appropriate. Regulatory authorization is required for all medical devices, regardless of device class. A potential exception to this requirement is for medical devices that were designed and constructed by local health care facility and staff for internal use. Similar to the United States, the SFDA requires post market surveillance to ensure safety and quality. This program is meant to be conducted by the Authorized Representative. With respect to the use of medical devices, it is the responsibility of the health care institution to inform the manufacturer and the SFDA of any adverse events associated with this use. We have appointed Al Sultan Saudi Medical Company as our responsible Authorized Representative for the KSA. Our Medical Device Marketing Authorization was renewed on July 26, 2016 and expires on May 14, 2020. In KSA it is possible for a foreign party to establish a Technical & Scientific Office and register the medical device, while working with a locally licensed Authorized Representative to conduct sales of such approved medical devices.
Kuwait
Medical devices in Kuwait are regulated by the Medicines and Medical Supplies, Pharmaceuticals and Herbal Medicines Registration and Control Administration Department in the Ministry of Health.
In order for any company/manufacturer to sell a medical device in Kuwait, the specific medical device must be approved for use and registered in Kuwait with the Ministry of Health. The manufacturer of the device, through its agent/distributor should submit an application to the Ministry of Health for the approval and registration of the device. The documents required to register a medical device with the Ministry of Health in summary include: (i) the original Manufacturing License and Good Manufacturing Practice certificates; (ii) the original Free Sale Certificate which should mention the trade name, scientific name, indications, and detailed composition for active and inactive ingredients and which should be issued by the health authority in the country of origin of the device; (iii) the status of registration of the product in the country of origin; (iv) the original letter of appointment of an exclusive agent/distributor for the device; (v) a list of countries where the product is registered with registration dates and numbers; (vi) a sample of the product with information about the product on the outer and inner packaging in English or Arabic (the information on the packaging should include: the name of the product, its content/composition, uses, batch number, manufacturing date, expiry date, storage conditions, and instructions on use); (vii) a certificate of analysis of the finished product; (viii) safety and efficacy studies from an approved international authority (and/or clinical studies if applicable); and (ix) any other information the Ministry of Health may require. Once all documents are in order and the Ministry of Health does not require any further information, it will register the device under the names of the manufacturer and the relevant agent/distributor.
The promotion, distribution and sale of medical devices in Kuwait can only be done by a Kuwaiti entity that is appointed by the manufacturer of the device as its exclusive agent/distributor for Kuwait. Such agent/distributor must be authorized by and registered with the Medicines and Medical Supplies, Pharmaceuticals and Herbal Medicines Registration and Control Administration Department in the Ministry of Health and the Ministry of Commerce and Industry to do so. The device may be sold in licensed pharmacies and other places approved by the Ministry of Health.
We have appointed Bader as our exclusive agent/distributor in Kuwait.
23
United Arab Emirates, or UAE
The most pertinent regulation is UAE Federal Law No. 4 of 1983 for the Pharmaceutical Profession and Institutions and to Medical Device Regulations. There are many similarities between the SFDA and the Registration and Drug Control Department that is run out of the Ministry of Health & Prevention of the UAE. Applications for registration of medical devices in the UAE are done with the UAE Ministry of Health Registration & Drug Control Department and must include data on effectiveness in addition to safety (a nod to the requirements of the FDA). The UAE body has its own device classification system that is most closely related to that used by the European Union, defined as class 1, low risk; class 2, medium risk but nonimplantable; class 3, medium risk but implantable; and class 4, high risk. The Obalon Balloon System is considered a Class 4 (high risk) device when delivered with a porcine-based gelatin capsule. We have appointed Sohail Faris Medical Equipment Trading as the responsible Authorized Representative for the UAE.
Privacy and Security Laws
There are numerous U.S. federal and state laws and regulations related to the privacy and security of personal information, including health information. Among others, the federal Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and their implementing regulations, (collectively referred to as HIPAA), establish privacy and security standards that limit the use and disclosure of protected health information, or PHI, and require covered entities and business associates to implement administrative, physical, and technical safeguards to ensure the confidentiality, integrity and availability of individually identifiable health information in electronic form, among other requirements.
Violations of HIPAA may result in civil and criminal penalties. Companies subject to HIPAA must also comply with HIPAA’s breach notification rule which requires notification of affected patients and the U.S. Department of Health and Human Services (HHS), and in certain cases of media outlets, in the case of a breach of unsecured PHI. The regulations also require business associates of covered entities to notify the covered entity of breaches by the business associate. State attorneys general also have the right to prosecute HIPAA violations committed against residents of their states, and HIPAA standards have been used as the basis for the duty of care in state civil suits, such as those for negligence or recklessness in misusing personal information. In addition, HIPAA mandates that HHS conduct periodic compliance audits of HIPAA covered entities and their business associates for compliance.
Many states in which we operate have laws that protect the privacy and security of sensitive and personal information, including health information, to which we are subject. These laws may be similar to or even more protective than HIPAA and other federal privacy laws. For example, the laws of the State of California, in which we operate, are more restrictive than HIPAA. California recently passed the California Consumer Privacy Act or CCPA, which will go into effect January 1, 2020. While certain health information we maintain may be exempt from the CCPA, other records and information we maintain on our customers may be subject to the CCPA. In certain cases, it may be necessary to modify our planned operations and procedures to comply with these state laws. Not only may some of these state laws impose fines and penalties upon violators, but also some may afford private rights of action to individuals who believe their personal information has been misused.
We may be subject to other state and federal privacy laws, including laws that prohibit unfair privacy and security practices and deceptive statements about privacy and security, laws that place specific requirements on certain types of activities, such as data security and texting, and laws requiring holders of personal information to maintain safeguards and to take certain actions in response to a data breach.
Anti-Kickback Statutes
The federal Anti-Kickback Statute prohibits persons from (among other things) knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in exchange for or to induce the referral of an individual, or the
24
recommending, furnishing or arranging for a good or service, for which payment may be made under a federal healthcare program such as Medicare or Medicaid.
Courts have interpreted the Anti-Kickback Statute quite broadly, holding that the statute will be violated if even one purpose of a payment – though not its sole or primary purpose – is to induce an act prohibited by the statute with a willful intent to act improperly. The statute prohibits many arrangements and practices that are otherwise lawful in businesses outside of the healthcare industry. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. Prosecutors may infer intent from the surrounding circumstances and, because courts have interpreted the statute to be violated if even one purpose of a payment is to induce the purchase of items or services paid for by federal healthcare programs, prosecutors have broad discretion in choosing arrangements to prosecute under the statute. There are statutory exceptions and regulatory “safe harbors” available to protect certain appropriately structured arrangements that otherwise would implicate the Anti-Kickback Statute. Those who structure their business arrangements to satisfy all of the criteria of a safe harbor are protected from liability under the statute. The failure of a transaction or arrangement to fit precisely within one or more safe harbors does not necessarily mean that it is illegal or that prosecution will be pursued. However, conduct and business arrangements that do not fully satisfy each applicable safe harbor may result in increased scrutiny by government enforcement authorities such as the Department of Health and Human Services Office of Inspector General.
Penalties for violation of the Anti-Kickback Statute are severe and may include, in addition to the fines of up to $100,000 per violation and imprisonment for up to ten years, penalties imposed under the Civil Monetary Penalties Law, or the CMP Law, including exclusion from participation in federal healthcare programs, civil monetary penalties of up to $100,000 for each improper act, and damages of up to three times the amount of remuneration at issue (regardless of whether some of the remuneration was for a lawful purpose). Because we do not anticipate that our products will be reimbursed by any federal healthcare program, we do not believe that we will be subject to the federal Anti-Kickback Statute.
Many states have adopted anti-kickback and self-referral laws similar to the Anti-Kickback Statute, however, and some of these state prohibitions may be broader in scope and apply to arrangements involving healthcare items or services reimbursed by any source, and not only by Medicare, Medicaid or another federal healthcare program. These state laws do not always have the same exceptions or safe harbors of the federal Anti-Kickback Statute. The business may be subject to some of these laws.
Government officials have focused recent enforcement efforts on the marketing of healthcare services and products, among other activities, and have brought cases against companies, and certain individual sales, marketing and executive personnel, for allegedly offering unlawful inducements to potential or existing customers in an attempt to procure their business.
False Claims Laws
The federal False Claims Act imposes liability on any individual or entity that, among other things, knowingly presents, or causes to be presented, a false or fraudulent claim for payment by a federal healthcare program. The qui tam or “whistleblower” provisions of the False Claims Act allow a private individual to bring actions on behalf of the federal government alleging that the defendant has violated the False Claims Act and to share in any monetary recovery. In recent years, the number of lawsuits brought against healthcare industry participants by private individuals has increased dramatically.
When an entity is determined to have violated the False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties of between $11,181 and $22,363 for each separate instance of false claim. As part of any settlement, the government may ask the entity to enter into a corporate integrity agreement, which imposes certain compliance, certification and reporting obligations. There are many potential bases for liability under the False Claims Act. Liability arises, primarily, when an entity knowingly submits, or causes another to submit, a false claim for reimbursement to the federal government, but also may arise when an entity knowingly makes a false statement material to an obligation to pay or transmit money or property to the federal government or knowingly conceals or knowingly and improperly avoids or decreases an obligation to pay or transmit money or property to the federal government. The federal government has used the False Claims Act to assert liability on the basis of inadequate care, kickbacks and other improper referrals, and the provision of inaccurate reimbursement coding advice, in addition to the more predictable allegations as to misrepresentations with respect to the services rendered. In addition, companies have been sued under the False Claims Act in connection with the
25
off-label promotion of products, for distributing products that fail to meet GMPs, and other sales and marketing practices. Moreover, the government may assert that a claim including items or services resulting from a violation of the Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act.
Various states have also enacted false claims and insurance fraud laws that are analogous to the federal False Claims Act. Many of these state laws apply to claims submitted to any third-party payor and are not limited to claims submitted to a federal healthcare program. The scope of these laws and the interpretations of them vary from state to state and are enforced by state courts and regulatory authorities, each with broad discretion. A determination of liability under such laws could result in fines and penalties and restrictions on a company’s ability to operate in these jurisdictions.
Transparency Laws
The federal Physician Payment Sunshine Act, or the Sunshine Act, which was enacted as part of the Patient Protection and Affordable Care Act, or the PPACA, generally requires certain manufacturers of a drug, device, biologic or other medical supply that is covered by Medicare, Medicaid or the Children’s Health Insurance Program and applicable group purchasing organizations to report on an annual basis: (i) certain payments and other transfers of value given to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals and (ii) any ownership or investment interest that physicians, or their immediate family members, have in their company. The payments required to be reported include the cost of meals provided to a physician, travel reimbursements and other transfers of value, including those provided as part of contracted services such as speaker programs, advisory boards, consultation services and clinical trial services. Under the statute, the federal government makes reported information available to the public. Failure to comply with the reporting requirements can result in significant civil monetary penalties ranging from $1,128 to $11,278 for each payment or other transfer of value that is not reported (up to a maximum per annual report of $169,170) and from $11,278 to $112,780 for each knowing failure to report (up to a maximum per annual report of $1.128 million). Additionally, there are criminal penalties if an entity intentionally makes false statements in the reports. Because we do not expect the Obalon Balloon System to be covered or reimbursed by any federal healthcare program, we do not believe that our business will be subject to the federal Sunshine Act.
There has been a recent trend of separate state regulation of payments and transfers of value by manufacturers of medical devices to healthcare professionals and entities, however, and some state transparency laws apply more broadly than does the federal Sunshine Act. There are also an increasing number of analogous state laws that require manufacturers to file reports with states on pricing and marketing information. Many of these laws contain ambiguities as to what is required to comply with the laws. For example, several states have enacted legislation requiring manufacturers to, among other things, establish and implement commercial compliance programs, file periodic reports with the state, make periodic public disclosures on sales, marketing, pricing, clinical trials and other activities and/or register their sales representatives. Certain state laws also regulate manufacturers’ use of physician and patient identifiable data. These laws may affect our sales, marketing and other promotional activities by imposing administrative and compliance burdens. In addition, given the lack of clarity with respect to these laws and their implementation, our reporting actions could be subject to the penalty provisions of the pertinent state and federal authorities. All of our activities are also potentially subject to federal and state consumer protection and unfair competition.
Other Federal Healthcare Fraud and Abuse Laws
We may also subject to other federal healthcare fraud and abuse laws, including HIPAA, which prohibits knowingly and recklessly executing a scheme or artifice to defraud any healthcare benefit program, including private payors, as well as knowingly and willfully falsifying, concealing or covering up a material fact by any trick, scheme or device or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. A violation of this statute is a felony and may result in fines, imprisonment or exclusion from government-sponsored programs. Similar to the federal Anti-Kickback Statute, a person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it in order to have committed a violation.
26
Licensing, Certification and Accreditation
Retail center construction and operation is subject to complex federal, state and local regulations relating to the adequacy of medical care, equipment, personnel, operating policies and procedures, fire prevention and compliance with building codes and environmental protection laws. Our planned retail centers may also be subject to periodic inspection by governmental and other authorities to assure continued compliance with the various standards necessary for licensing and accreditation.
Corporate Practice of Medicine and Fee Splitting
Some of the states in which we plan to operate our retail centers have laws that prohibit unlicensed persons or business entities, including corporations, from employing physicians and also laws that prohibit certain direct or indirect payments or fee-splitting arrangements between physicians and unlicensed persons or business entities. These laws are intended to prevent unlicensed persons from interfering with or influencing the physician’s professional judgment. Activities other than those directly related to the delivery of healthcare may be considered an element of the practice of medicine in many states. Possible sanctions for violations of these restrictions include loss of a physician’s license, civil and criminal penalties and rescission of business arrangements that may violate these restrictions. These statutes vary from state to state, are often vague and seldom have been interpreted by the courts or regulatory agencies. Although we intend to exercise care to structure our arrangements with physicians to comply with the relevant state law, we cannot assure you that governmental officials responsible for enforcing these laws, or other third parties, will not assert that we, or transactions in which we are involved, are in violation of such laws, or that such laws ultimately will be interpreted by the courts in a manner consistent with our interpretations.
Foreign Corrupt Practices Act
The Foreign Corrupt Practices Act, or FCPA, prohibits U.S. businesses and their representatives from offering to pay, paying, promising to pay or authorizing the payment of money or anything of value to a foreign official in order to influence any act or decision of the foreign official in his or her official capacity or to secure any other improper advantage in order to obtain or retain business. The FCPA also obligates companies whose securities are listed in the United States to comply with accounting provisions requiring us to maintain books and records, which in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the corporation, including international subsidiaries, if any, and to devise and maintain a system of internal accounting controls sufficient to provide reasonable assurances regarding the reliability of financial reporting and the preparation of financial statements. The scope of the FCPA includes interactions with certain healthcare professionals in many countries.
International Laws
In Europe, and throughout the world, other countries have enacted anti-bribery laws and/or regulations similar to the FCPA. Violations of any of these anti-bribery laws, or allegations of such violations, could have a negative impact on our business, results of operations and reputation.
There are also international privacy laws that impose restrictions on the access, use, and disclosure of health information. All of these laws may impact our business. Our failure to comply with these privacy laws or significant changes in the laws restricting our ability to obtain required patient information could significantly impact our business and our future business plans.
U.S. Healthcare Reform
Changes in healthcare policy could increase our costs and subject us to additional regulatory requirements that may interrupt commercialization of the Obalon Balloon System. By way of example, PPACA substantially changed the way healthcare is financed by both governmental and private insurers, and significantly impacted the medical device industry. PPACA, among other things, imposed a 2.3% excise tax on any entity that manufactures or imports medical devices offered for sale in the
27
United States, with limited exceptions. Although the excise tax was suspended from 2016 through 2019, absent further legislative action, the tax will be reinstated starting January 1, 2020.
There will continue to be proposals by legislators at both the federal and state levels, regulators and third-party payors to reduce costs while expanding individual healthcare benefits. Certain of these changes could impose additional limitations on the prices we will be able to charge and/or patients’ willingness to pay for the Obalon Balloon System. While in general it is too early to predict what effect, if any, PPACA and its implementation, or any future healthcare reform legislation or policies will have on our business, current and future healthcare reform legislation and policies could have a material adverse effect on our business and financial condition.
Employees
As of June 14, 2019, we had 37 full-time employees, and eight temporary employees. These included eight in manufacturing and operations, seven in sales and marketing, eight in research and development, eight in clinical affairs, regulatory affairs and quality assurance and six in finance, general administrative and executive administration. None of our employees are represented by a labor union or are parties to a collective bargaining agreement, and we believe that our employee relations are good.
28